Le séquençage du génome
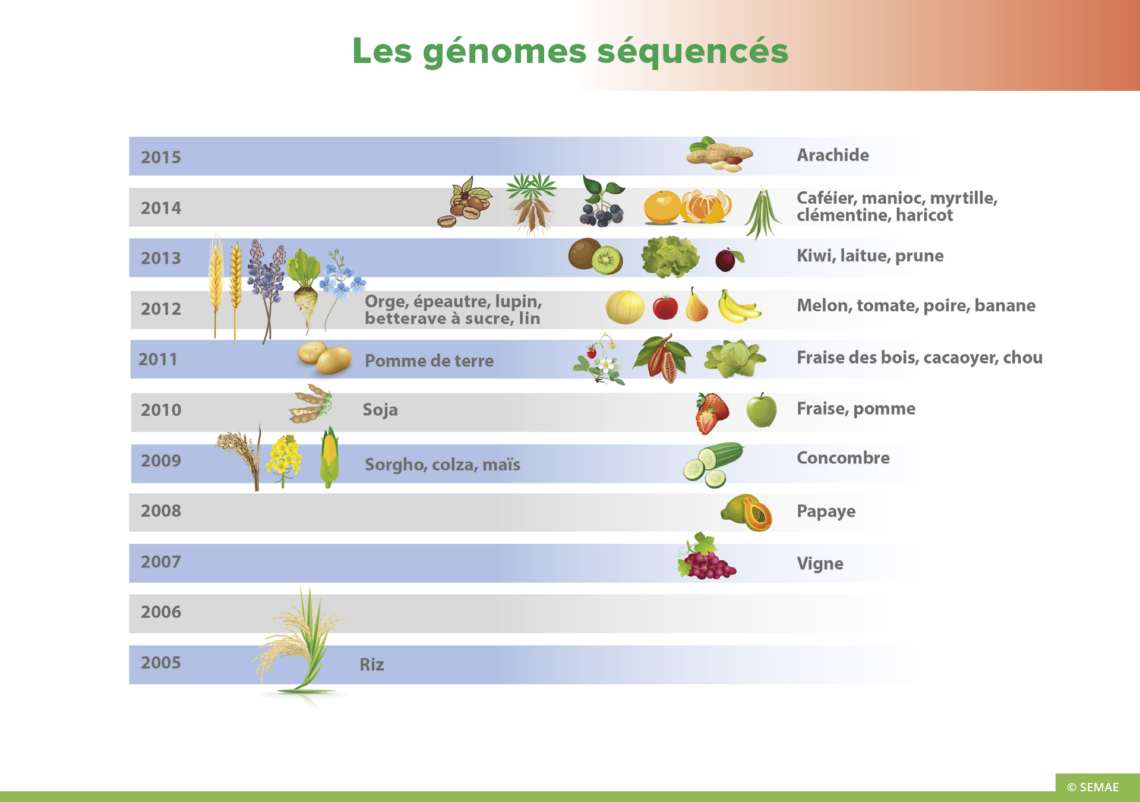
AUTOMATISATION DU SÉQUENÇAGE
La très grande majorité des séquençages réalisés se font de manière automatique en utilisant le même principe d’hybridation moléculaire et la machinerie cellulaire qui permet d’amplifier des fragments d’ADN (PCR), mais en utilisant désormais des marqueurs fluorescents et la chromatographie pour déterminer la taille des fragments d’ADN étudiés et lire les quatre nucléotides d’un fragment.
Le séquençage se fait (comme pour la PCR) avec l’ADN matrice du gène à étudier, une amorce, des enzymes particulières : les ADN polymérases qui sont capables de synthétiser un brin complémentaire d’ADN, des nucléotides (A,T,C,G), mais aussi dans ce type de séquençage avec des didésoxynucléotides fluorescents qui diffèrent des nucléotides par l’absence d’un groupement OH et qui empêchent l’ADN polymérase de continuer la synthèse du brin complémentaire. Quatre fluorochromes différents sont utilisés, un par nucléotide.
Lors de l’hybridation moléculaire, quand l’ADN polymérase ajoute un didésoxynucléotide fluorescent sur le brin en cours de synthèse au lieu d’un nucléotide, le brin s’interrompt. Au final, de nombreux brins sont obtenus, de taille variable mais tous terminés par un didésoxynucléotide fluorescent. Par automatisation du tri des brins obtenus en fonction de leur taille, la séquence peut être lue par le séquenceur selon la fluorescence observée.
La combinaison des marqueurs
Il est rare d’avoir besoin d’information sur un seul locus d’un génome.
Pour détecter des QTL (Quantitative Trait Locus, petite région du génome dont la variation modifie un caractère), il est nécessaire de baliser l’ensemble du génome à l’aide d’un grand nombre de marqueurs : il faut qu’au moins un de ces marqueurs soit suffisamment proche du QTL pour qu’on observe une liaison génétique entre le marqueur et le QTL. Pour cela, différentes approches nouvelles permettent de tester plusieurs de marqueurs simultanément tout en réduisant les temps de manipulation et leur coût.
L’étude de polymorphisme portant sur un nucléotide
Les SNP (Single Nucleotide Polymorphism, simple substitution nucléotidique) constituent la forme la plus abondante de variations génétiques : c’est un type de polymorphisme de l’ADN dans lequel deux chromosomes diffèrent sur un segment donné par une seule paire de bases. Ces SNP sont stables, très abondants et distribués uniformément dans tout le génome.
Différentes techniques permettent de mettre en évidence des différences d’un nucléotide (RFLP, RAPD, AFLP, SSR…) ou encore les microbilles universelles mais une des plus performantes actuellement est l’usage des puces à ADN car elle présente un potentiel d’automatisation supérieur et peut être réalisée à très haut débit.
Les microbilles sont recouvertes de centaines de milliers de molécules d’un oligonucléotide spécifique (caractéristique d’un locus) et sont identifiables par un code barre.
Cette technique permet de visualiser et donc d’identifier quelques centaines de SNP par échantillon.
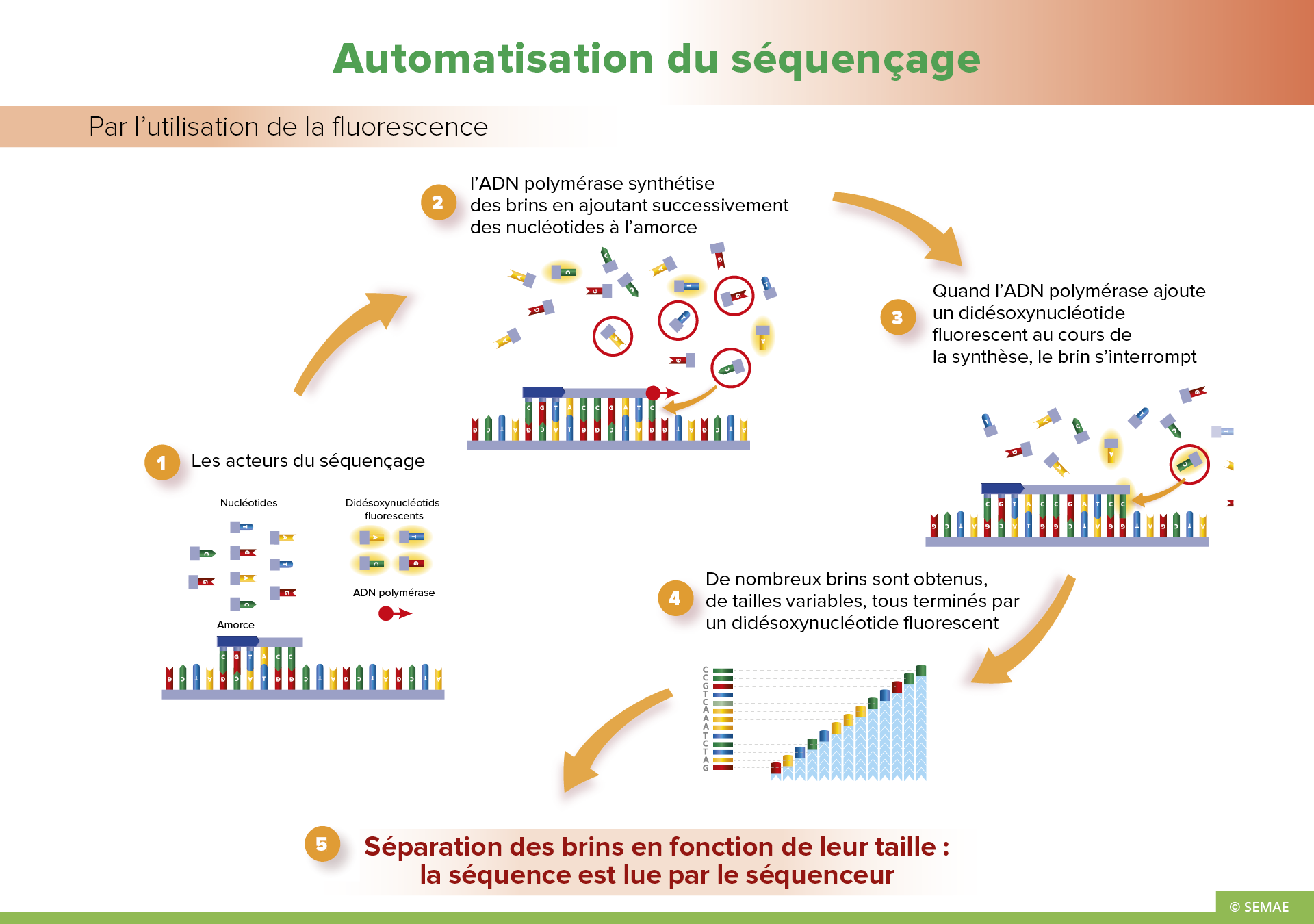
LES GÉNOMES SÉQUENCÉS
Le premier génome de plante entièrement séquencé et publié fut celui de l’espèce modèle de laboratoire Arabidopsis thaliana en 2000. Depuis, de nombreuses plantes ont été séquencées partiellement ou totalement. Ces séquençages ont été rendus possibles par la diminution des coûts du génotypage de grands nombres d’individus pour réaliser des cartes servant à assembler les différents fragments de séquence entre eux. Plus récemment, de nouvelles techniques de séquençage dites de « nouvelle génération » ont énormément réduit le coût par fragment séquencé, mais posent de gros problèmes d’analyse informatique pour l’assemblage des millions de tout petits morceaux de séquence obtenus, afin de reconstituer un génome complet.
La plupart des génomes des grandes cultures sont séquencées
- Riz (2005)
- Maïs, sorgho, colza (2009)
- Soja (2010)
- Pomme de terre (2011)
- Orge (2012)
- Épeautre (2013)
- Lupin (2013)
- Haricot (Phaseolus vulgaris) (2014)
- Betterave à sucre (2014)
- Arachide (2015)
- Tournesol (2016)
- Reste le blé…
Beaucoup de fruits et légumes ont aussi été séquencés
- Vigne (2007)
- Papaye (2008)
- Concombre (2009)
- Fraise, pomme, cacao (2010)
- Choux (2011)
- Tomate, melon, poire, banane (2012)
- Orange (citrus) (2014)
- Laitue, prune : en cours
Evolution du nombre de génomes publiés
En avril 2016, 800 animaux, 248 plantes, 1.450 champignons, 8.288 bactéries et 5.396 virus sont ainsi séquencés.
La plupart des génomes sont disponibles sur Internet. Une synthèse des publications et les références des consortiums de génotypage sont également accessibles en ligne. Exemple :
U.S. National Center for Biotechnology Information
De plus, au sein d’une espèce, de nombreuses variétés peuvent être cartographiées. Cette connaissance fine des génomes est possible grâce aux nouveaux outils moléculaires et informatiques qui permettent d’analyser de nombreuses données à des coûts de plus en plus faibles.
Enrichir l’amélioration des plantes
Les attentes vis-à-vis de l’amélioration variétale sont de plus en plus diversifiées. Mais les travaux de recherche exigent de nombreuses années. L’un des objectifs est donc de réduire la durée et les coûts de la sélection, en particulier par la fixation plus rapide des caractères acquis. L’haplo diploïdisation est l’une des techniques largement utilisée pour de nombreuses espèces. D’autres techniques permettent également de réduire la durée de création: mutagénèse dirigée, transgénèse, cisgénèse… Ces techniques font appel aux outils de culture in vitro réalisée en laboratoire et donc évitant toute contamination bactérienne ou virale.
La création variétale permet l’acquisition par les plantes de caractères intéressants présents au sein de l’espèce ou dans des espèces voisines. Les croisements inter spécifiques sont utilisés mais aussi des techniques qui augmentent l’accès à la diversité génétique. La connaissance des génomes et de leur fonctionnement ont en effet donné naissance à de nouveaux outils qui agissent directement sur le génome (ajout, modification ou inhibition de gènes) ou via des protéines qui interfèrent avec le génome. Toutes ces techniques ouvrent ainsi sur des domaines d’application de plus en plus larges.